

Atoms are quantum systems with a positively charged nucleus and negatively charged electrons. Simulating the interactions within molecules formed by multiple atoms is a significant challenge in modern science. Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin, in collaboration with Google DeepMind, have developed a new machine learning (ML) algorithm that enables accurate simulations of single or multiple molecules over long time scales. This breakthrough, published in Nature Communications, has potential applications in drug development and material design.
Traditional methods for simulating molecular dynamics rely on solving the Schrödinger equation, which is computationally expensive, particularly for molecules with many atoms. These methods require solving the equation thousands or millions of times, quickly exceeding available computational resources.
“The simulation of such interactions and the resulting predictions for complex processes like protein folding or the binding between individual molecules is a long-held dream of many chemists and material scientists, and would save many expensive and labor-intensive experiments,” said BIFOLD researcher Thorben Frank.
Recent advances in machine learning offer a solution by predicting the outcome of electronic interactions without explicitly solving the Schrödinger equation, reducing computational costs. However, incorporating physical system invariances into ML models has traditionally been costly and limited the speed of simulations.
The BIFOLD researchers addressed this by creating an algorithm that decouples invariances from other chemical information from the outset. This approach simplifies the learning process and allows the ML model to focus on the most complex physical information, significantly reducing computational costs.
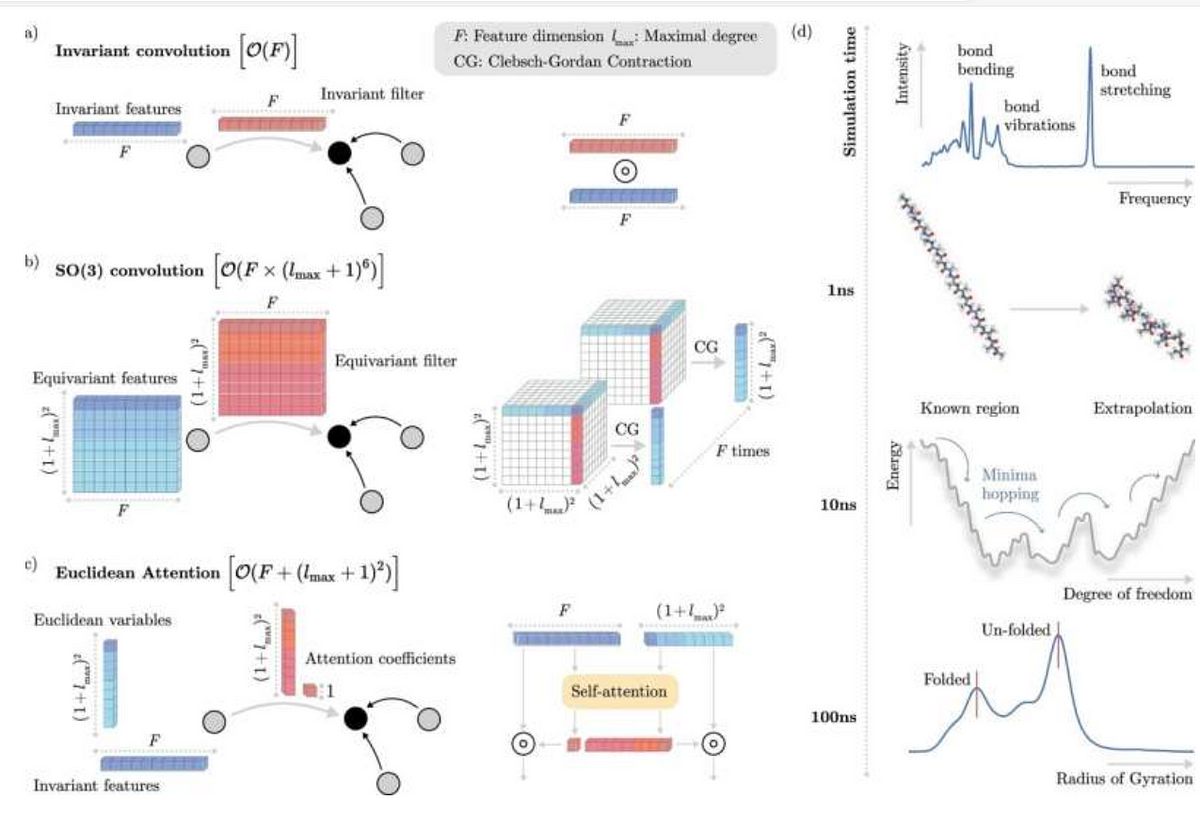 a Illustration of an invariant convolution. b Illustration of an SO(3) convolution. c Illustration of the Euclidean attention mechanism that underlies the SO3krates transformer. We decompose the representation of molecular structure into high dimensional invariant features and equivariant Euclidean variables (EV), which interact via self-attention. d The combination of simulation stability and computational efficiency of SO3krates enables the analysis of a broad set of properties (power spectra, folding dynamics, minima analysis, radius of gyration) on different simulation timescales.
a Illustration of an invariant convolution. b Illustration of an SO(3) convolution. c Illustration of the Euclidean attention mechanism that underlies the SO3krates transformer. We decompose the representation of molecular structure into high dimensional invariant features and equivariant Euclidean variables (EV), which interact via self-attention. d The combination of simulation stability and computational efficiency of SO3krates enables the analysis of a broad set of properties (power spectra, folding dynamics, minima analysis, radius of gyration) on different simulation timescales.
Dr. Stefan Chmiela, a BIFOLD researcher who led the project, explained that simulations previously requiring months or even years on high-performance computer clusters can now be completed within a few days on a single computer node. This significant improvement in efficiency allows for long-time scale simulations, which are essential for understanding the structure, dynamics, and functioning of atomistic systems, providing deeper insights into complex and fundamental natural processes.
Prof. Dr. Klaus-Robert Müller, BIFOLD co-director and Principal Scientist at Google DeepMind, highlighted the potential of combining advanced machine learning techniques with physical principles to address longstanding challenges in computational chemistry. He emphasized that this research continues a critical line of inquiry focused on scaling machine learning approaches for realistic chemical systems of practical interest.
Dr. Oliver Unke, meanwhile, a Senior Research Scientist at Google DeepMind, added that while earlier efforts had succeeded in scaling models to thousands of atoms, new advancements like this one could enable simulations involving even larger numbers of atoms.
The new algorithm could allow researchers to simulate molecular interactions with proteins, aiding drug development without costly experiments. The team demonstrated the algorithm’s potential by identifying the most stable version of docosahexaenoic acid, a critical fatty acid in the human brain, a task previously infeasible with traditional methods.
Future algorithms will need to handle even larger structures with millions of atoms, requiring accurate descriptions of complex, long-range physical interactions.



